![]()
Últimos números

Último Número. Vol.30, Nº 2 2023
Tumores del sistema nervioso central en niños. Experiencia del hospital infantil Reina Sofía.
Revista:
Volumen 21. 2014. Nº 1
Tipo de artículo:
Originales Resumen:
Antecedentes: Los tumores del sistema nervioso central constituyen la segunda neoplasia más frecuente en niños y la primera causa de muerte por enfermedad oncológica en la infancia.
Objetivos: Presentar la casuística de los tumores del sistema nervioso central en niños del Hospital Universitario Reina Sofía y contrastar los datos con la literatura publicada.
Métodos: Estudio retrospectivo de los pacientes diagnosticados y/o tratados en la Unidad de Oncología Pediátrica del Hospital Reina Sofía durante un período de 10 años.
Resultados: Se obtuvieron 74 casos con una relación varón/mujer 0,51/0,48 y una edad media al diagnóstico de 8,8 años. El 99% de los casos correspondieron a un tumor primario. Un caso fue secundario a la exposición previa a radiaciones ionizantes. La cefalea fue la forma clínica más frecuente de presentación, excepto en menores de 4 años en los que predominaron las alteraciones de la marcha. Los gliomas de bajo grado representaron la variedad histológica más común (35,13%) y en cuanto a la localización, predominaron los supratentoriales (61%). Recibieron tratamiento quirúrgico 52 pacientes. La supervivencia en nuestra serie es del 87,83% a los 5 años del diagnóstico.
Conclusiones: Los tumores del sistema nervioso central infantiles suelen tener un origen primario. Los gliomas de bajo grado constituyen la variedad histológica más frecuente y la cefalea es la forma de presentación más común. En nuestra serie predominó la localización supratentorial. El tratamiento de elección es la cirugía. La indicación de radioterapia, quimioterapia o agentes biológicos depende tipo de tumor, la localización y edad del paciente.
Central nervous system tumors in children. Reina sofia children’s hospital experience.
Background: Tumors of the Central nervous system are the second most common malignancy in children and the leading cause of death from cancer disease in childhood.
Objectives: To present the casuistry central nervous system tumors in children of Reina Sofia University Hospital and compare the data with the published literature.
Methods: Retrospective study of patients diagnosed and / or treated at the Pediatric Oncology Unit of Reina Sofia Hospital for a period of 10 years.
Results: 74 cases with a male / female 0.51 / 0.48 ratio and mean age at diagnosis 8.8 years were obtained. 99% of the cases were primary tumor. One case was secondary to previous exposure to ionizing radiation. Headache was the most common clinical presentation , except for children under 4 years that predominated gait disturbances . The low-grade gliomas represent the most common histological subtype (35.13 %) and the location , mainly supratentorial ( 61%). 52 patients received surgical treatment . Survival in our series is 87.83 % at 5 years after diagnosis.
Conclusions: Children central nervous system tumors usually have a primary origin. Low-grade gliomas are the most common histological variety and headache is the most common form of presentation. In our series, the supratentorial location predominated . The treatment of choice is surgery. The indication for radiotherapy, chemotherapy or biological agents depends on tumor type, location, and patient age.
10
S
OCIEDAD
DE
P
EDIATRÍA
DE
A
NDALUCÍA
O
CCIDENTAL
Y
E
XTREMADURA
Volumen XXI Nº 1 Mayo 2014
Introducción
Los tumores del sistema nervioso central (SNC) son
un grupo de entidades tanto benignas como malignas
que afectan al encéfalo y la médula espinal
(1)
. Cons-
tituyen la segunda neoplasia más frecuente en niños,
después de la leucemia, y por consiguiente el tumor
de órgano sólido más común en la infancia. Represen-
tan entre un 15 y 20% del total de los casos nuevos
de cáncer que se diagnostican por año en España en
menores de 15 años y la primera causa de muerte por
enfermedad oncológica a esta edad
(1, 2, 3)
.
En la infancia, la incidencia anual de este grupo de
tumores es de 2,8 casos por cada 100.000 niños
(2)
.
La tasa de incidencia se ha incrementado desde la dé-
cada de los 90, lo que se atribuye a la disponibilidad y
sensibilidad del diagnóstico por imagen mediante reso-
nancia magnética
(4)
.
Según el Registro Nacional de Tumores Infantiles de
la Sociedad Española de Hematología y Oncología Pe-
diátricas (RNTI-SEHOP), atendiendo a los datos de in-
cidencia de base poblacional, de 4439 casos de tu-
mores diagnosticados en el grupo de edad de 0 a 14
años, entre 2000 y 2012, 953 son tumores del SNC
(5)
.
La etiología es desconocida en la mayoría de los ca-
sos. No obstante, se han identificado distintos factores
implicados en la carcinogénesis de tumores determi-
nados. Entre ellos, el mejor conocido son las radiacio-
nes ionizantes, en relación con el desarrollo de gliomas,
meningiomas o schawannomas
(6)
. Igualmente, predis-
ponen al desarrollo de tumores del SNC ciertos sín-
dromes genéticos, como la neurofibromatosis tipo 1,
asociada al desarrollo de neurofibromas, gliomas del
nervio óptico o astrocitomas; y la esclerosis tuberosa,
que se asocia al astrocitoma subependimario de célu-
las gigantes. Algunas inmunodeficiencias congénitas y
la presencia de otros tumores como la adenomatosis
endocrina múltiple, el retinoblastoma trilateral o el tu-
mor rabdoide maligno renal se han relacionado con la
aparición de tumores cerebrales
(7)
.
La clínica de presentación va a depender de la loca-
lización del tumor principalmente, pero también de la
edad y de la velocidad de crecimiento de la neoplasia
(2,3,4,8)
. Los signos y síntomas pueden ser secundarios a
la obstrucción de la circulación del líquido cefalorraquí-
deo (LCR) o a la compresión o infiltración de estructu-
ras adyacentes por el tumor
(4,8)
. En lactantes y niños
pequeños la hipertensión intracraneal puede cursar de
forma más insidiosa dada la elasticidad del cráneo al
encontrarse abierta la fontanela y separadas las sutu-
ras. La tensión de la fontanela y el perímetro craneal
pueden servir de signo guía en estos casos
(4)
.
En cuanto a la localización, se distinguen dos gran-
des grupos según el tumor se encuentre por encima o
por debajo de la tienda del cerebelo, siendo los infra-
tentoriales los más numerosos (55%), salvo en lactan-
tes en los en los que predominan los supratentoriales
(3)
.
Desde el punto de vista histológico, los gliomas de
bajo grado constituyen el grupo más numeroso de tu-
mores del SNC en niños seguidos de los tumores em-
brionarios como el meduloblastoma
(1,2)
.
En lo referente al tratamiento, la resección tumoral
completa o lo más amplia posible constituye la primera
opción terapéutica en la mayoría de los casos, con el
objetivo de confirmar el diagnóstico y reducir el volu-
men tumoral
(3)
. La histología, y la localización en el caso
de tumores inextirpables, determinarán la indicación de
radioterapia con/sin quimioterapia. Agentes biológicos
tipo anticuerpos monoclonales o inhibidores del m-TOR,
podrían tener una eficacia igual o incluso superior a la
de la radioterapia y quimioterapia convencional, con una
mayor penetrancia en el SNC y una menor toxicidad
(9)
.
A continuación presentamos la casuística de nues-
tro Centro.
Material y Métodos
Realizamos un estudio descriptivo retrospectivo de
los niños y adolescentes diagnosticados y/o tratados
de tumores del SNC en la Unidad de Oncología Pediá-
trica del Hospital Universitario Reina Sofía de Córdoba
entre Febrero de 2003 y Octubre de 2013.
Se analizaron las siguientes variables: género y
edad al diagnóstico, si se trata de un tumor primario o
secundario, la existencia de patología predisponente,
la forma clínica de presentación, el tiempo de demora
diagnóstica desde el debut de los síntomas, la locali-
zación primaria del tumor, el tratamiento recibido y la
supervivencia.
Resultados
Se incluyen en el estudio 74 pacientes con una rela-
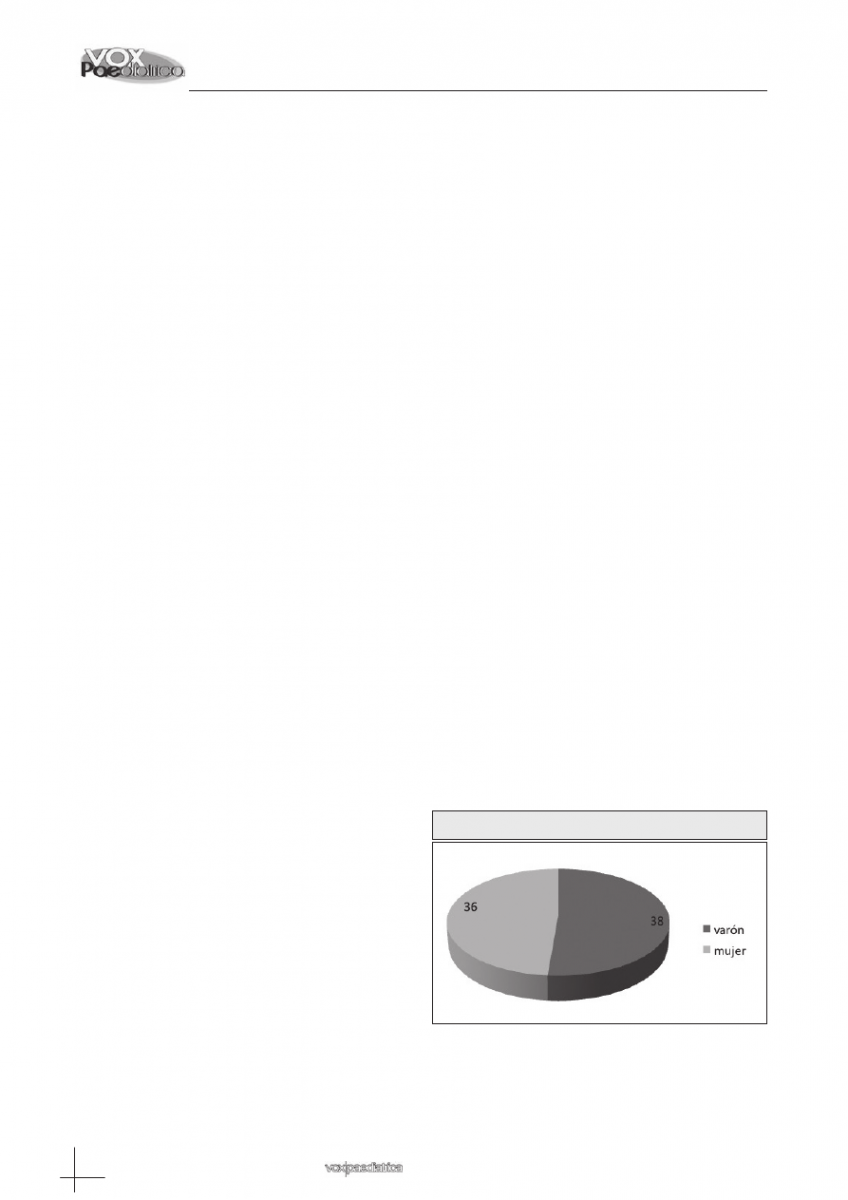
ción varón/mujer 0.51/0.48 (figura 1). La edad media
al diagnóstico fue de 8.8 años, con un intervalo que
osciló entre los 2 días de vida a los 18 años y 8 meses
al diagnóstico.
Figura 1: Distribución por género.
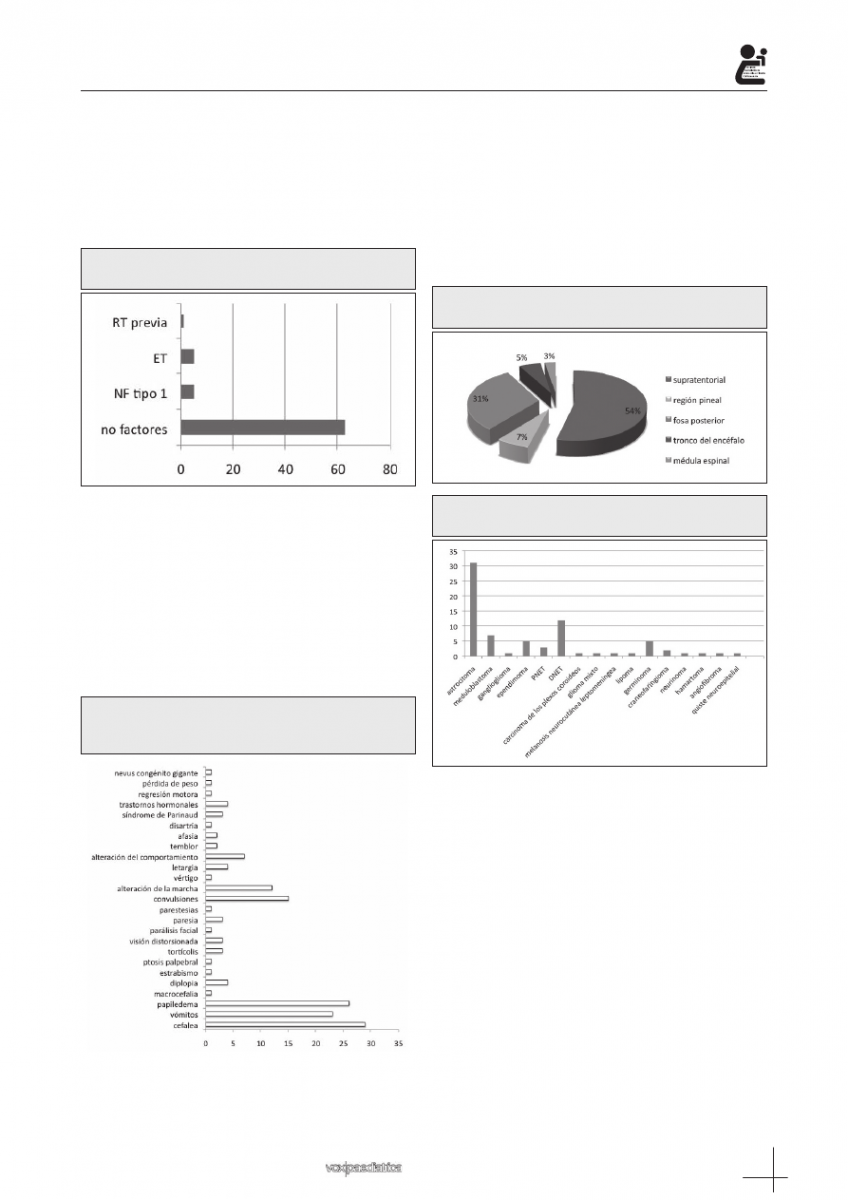
Patología predisponente: El 99% de los tumores
fueron primarios, tan sólo se registró un tumor cere-
bral secundario con el antecedente de exposición a ra-
diaciones ionizantes. En cuanto a patología predispo-
nente, cinco pacientes habían sido diagnosticados de
11
Volumen XXI Nº 1 Mayo 2014
B
AENA
-G
ÓMEZ
M.A.
ET
AL
- T
UMORES
DEL
SISTEMA
NERVIOSO
CENTRAL
EN
NIÑOS
. E
XPERIENCIA
DEL
HOSPITAL
INFANTIL
REINA
SOFÍA
esclerosis tuberosa y otros cinco de neurofibromatosis
tipo 1 (figura 2). Los cinco primeros presentaron as-
trocitomas subependimarios de células gigantes. Dos
de los pacientes con neurofibromatosis tipo 1 desarro-
llaron gliomas del nervio óptico y tres astrocitomas de
bajo grado, uno infratentorial y dos supratentoriales.
Figura 2: Patología predisponerte. 63 pacientes no
presentaban factores predisponentes.
Clínica de presentación: Los signos y síntomas al
diagnóstico se detallan en la figura 3. Un subgrupo de
16 pacientes fueron menores de 4 años al diagnóstico.
En este subgrupo predominaron las alteraciones de la
marcha como forma clínica de presentación. Por su
parte, la cefalea fue el síntoma más frecuente en los pa-
cientes diagnosticados de neurofibromatosis tipo 1. La
mediana del tiempo transcurrido desde el debut de los
síntomas hasta el diagnóstico fue de 1,5 meses en el
conjunto de los pacientes globalmente y de 0,5 meses
en menores de 4 años.
Figura 3: Signos y síntomas al diagnóstico de los
tumores del sistema nervioso central en niños del
Hospital Reina Sofía.
Localización: La localización tumoral fue funda-
mentalmente supratentorial (figura 4).
Histología: El astrocitoma fue la variedad histológica
más frecuente (41,89%), concretamente el astrocitoma
de bajo grado con 31 casos (35,13%), seguido del tu-
mor disembrioblástico del neuroepitelio (DNET), 12 ca-
sos (16,21%) y del meduloblastoma, 7 casos (9,45%).
Cinco pacientes (6,75%) se diagnosticaron de germi-
noma, cinco (6,75%) de ependimoma, tres (4,05%) de
tumor neuroectodérmico primitivo (PNET) y dos (2,7%)
de craneofaringioma. Otras variedades histológicas ob-
jetivadas se presentan en la figura 5.
Figura 4: Localización de los tumores del sistema
nervioso central en niños.
Figura 5: Variedades histológicas de los tumores de
los tumores del sistema nervioso central en niños.
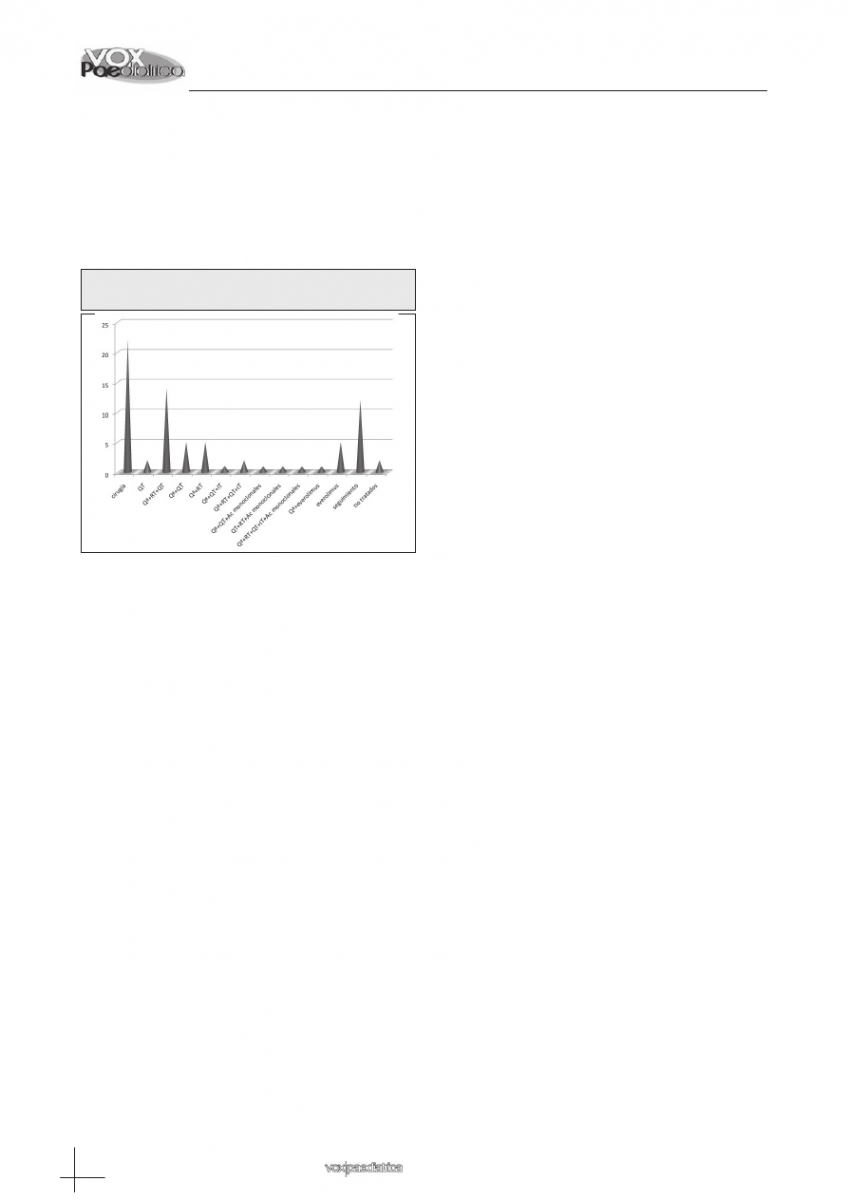
Tratamiento: Un total de 52 (70,27%) pacientes se
sometieron a cirugía. Se realizó extirpación completa
del tumor en un 53,84% de los casos y parcial en un
38,46%. En el primer caso el astrocitoma de bajo grado
fue la histología predominante, mientras que en el se-
gundo el germinoma. En 4 pacientes (7,69%) con diag-
nóstico anátomo-patológico de astrocitoma anaplá-
sico, ganglioglioma de alto grado, astrocitoma pilocí-
tico y germinoma, tan sólo se realizó biopsia. Veintidós
(42.3%) fueron subsidiarios de tratamiento quirúrgico
sin necesidad de terapia adyuvante, siendo en este
caso la histología más frecuente el DNET. Veintisiete
(36,48%) recibieron quimioterapia. Veintiún pacientes
(28,37%) recibieron radioterapia, 11 de ellos (14,86%)
local y 10 (13,51%) craneoespinal. Tres pacientes
(2,70%), afectos de glioblastoma multiforme, medulo-
blastoma y glioma difuso del tronco del encéfalo, pre-
cisaron la administración de anticuerpos monoclonales
(bevacizumab y/o nimotizumab) además de la terapia
tradicional. Cinco pacientes (6,75%) diagnosticados de
astrocitoma subependimario de células gigantes en el
12
S
OCIEDAD
DE
P
EDIATRÍA
DE
A
NDALUCÍA
O
CCIDENTAL
Y
E
XTREMADURA
Volumen XXI Nº 1 Mayo 2014
contexto de esclerosis tuberosa, recibieron tratamiento
con inhibidores del m-TOR. Un lactante diagnosticado
de carcinoma de plexos coroideos fue derivado a otro
centro de referencia para tratamiento por neurocirugía,
y una paciente diagnosticada de un tumor infiltrante de
tronco del encéfalo rechazó la quimioterapia paliativa
propuesta (figura 6).
Figura 6: Tratamiento tumores del sistema nervioso
central en niños.
Ac: anticuerpos; IT: intratecal; Qª: cirugía; QT: quimioterapia;
RT: radioterapia.
Supervivencia: La supervivencia a los 3 años en
nuestra serie es del 90,54% y del 87,83% a los 5 años.
El tipo histológico asociado a una mayor supervivencia
es el astrocitoma de bajo grado en ambos casos y la lo-
calización supratentorial. El 1,5 % de los pacientes reci-
bió quimioterapia intratecal, anticuerpos monoclonales
o inhibidores del m-TOR además del tratamiento quirúr-
gico asociado a quimioterapia sistémica y/o radiotera-
pia. Por su parte, se trataron con everolimus el 7,7%.
De los pacientes que sobrevivieron a los 3 años del
diagnóstico, el 31,3% se había sometido a tratamiento
quirúrgico. Se realizó resección completa en el 37,3%,
parcial en el 28,4% y biopsia en el 6%. El 17,9% reci-
bió cirugía con radioterapia y quimioterapia sistémica y
7,5% cirugía con una de las dos modalidades terapéu-
ticas previamente mencionadas. De los pacientes que
sobrevivieron a los 5 años del diagnóstico, el 32,3%
se había sometido a tratamiento quirúrgico. Se realizó
resección completa en el 38,5%, parcial en el 26,2% y
biopsia en el 6,2%. El 16,9% recibió cirugía con radio-
terapia y quimioterapia sistémica y 7,7% cirugía con
una de las dos modalidades terapéuticas previamente
mencionadas.
Han fallecido 9 pacientes, cuya supervivencia media
fue de 20,31 meses con un intervalo que osciló entre
los 0,56 y 45,96 meses desde el diagnóstico y una me-
diana de edad de 11 años.
Discusión
Los tumores del SNC constituyen la neoplasia de
órgano sólido más común en la infancia
(1,2)
. Presentan
un primer pico de incidencia en la primera década de la
vida y un segundo entre los 70 y 80 años de edad, con
un ligero predominio en varones y raza caucásica
(1,2,3)
.
Según los datos del RNTI-SEHOP, entre 1980 y
2012 los tumores del SNC fueron el segundo grupo de
neoplasias más numeroso tras las leucemias, con 4605
casos sobre un total de 22938 tumores. El 96,5 % se
diagnosticó en menores de 14 años y el 3,4 % en ado-
lescentes de 15 a 19 años de edad. En ambos grupos
de edad la frecuencia en varones fue mayor, 55,8% y
60,5% respectivamente
(5)
.
En nuestra serie también hubo un ligero predominio
de la incidencia en varones, 0,51:0,48, y en la primera
década de la vida.
Los tumores cerebrales secundarios son aquellos
que aparecen años después del tratamiento del tu-
mor primario y cuya histología es diferente
(10)
. El estu-
ido de Neglia y col. incluyó 14361 pacientes diagnosti-
cados de distintos tumores primarios (leucemia, tumor
del SNC, linfoma Hodgkin y no Hodgkin, tumor renal,
neuroblastoma, sarcoma de partes blandas y sarcoma
óseo), con una supervivencia de al menos 5 años
(11)
.
Del total de pacientes, 166 desarrollaron un tumor se-
cundario en el SNC, siendo los más frecuentes los
meningiomas (39.76%) y los gliomas (24%). El tiempo
desde el diagnóstico del tumor primario osciló entre 5
y 28 años, con una mediana de 14 años. Los gliomas
debutaron más precozmente que los meningiomas.
Los tumores primarios más frecuentes fueron la leuce-
mia aguda linfoblástica, en relación con el desarrollo de
gliomas, y los tumores primarios del SNC en relación
en los meningiomas. Como factor de riesgo más im-
portante se identificaron las radiaciones ionizantes. En
nuestra serie, tan sólo 1 caso fue un tumor secundario.
Se trata un varón con antecedente de leucemia aguda
linfoblástica diagnosticada a los 4 años de edad, que
recidivó en SNC a los 3 años del diagnóstico recibiendo
entonces radioterapia corporal total previa al trasplante
de médula ósea. A los 10 años del trasplante presentó
un glioma anaplásico (grado III de la OMS). El paciente
recibió quimioterapia paliativa y falleció a los 5 meses
del diagnóstico.
El 99% restante fueron tumores primarios. Un 13,5%
estaba diagnosticado de un síndrome genético con
predisposición al desarrollo de tumores del SNC, lo que
supone un total de 10 pacientes. Pinho y col. anali-
zaron retrospectivamente las características epidemi-
ológicas de los tumores primarios del SNC diagnosti-
cados en un grupo de 741 pacientes menores de 21
años de edad
(12)
. Observaron que el 4% de estos pa-
cientes asociaba un síndrome genético. La neurofibro-
matosis tipo 1 fue el síndrome más frecuente (48.27%),
seguido de la neurofibromatosis tipo 2 (24,13%) y la
esclerosis tuberosa (20,68%). Menos frecuentes fueron
el síndrome de Li-Fraumeni (3.44%) y el síndrome de
Gorlin (3.44%). Al igual que en nuestra serie, la neurofi-
bromatosis tipo 1 se asociaba al desarrollo de astroci-
13
Volumen XXI Nº 1 Mayo 2014
B
AENA
-G
ÓMEZ
M.A.
ET
AL
- T
UMORES
DEL
SISTEMA
NERVIOSO
CENTRAL
EN
NIÑOS
. E
XPERIENCIA
DEL
HOSPITAL
INFANTIL
REINA
SOFÍA
tomas de bajo grado y la esclerosis tuberosa a los as-
trocitomas subependimarios.
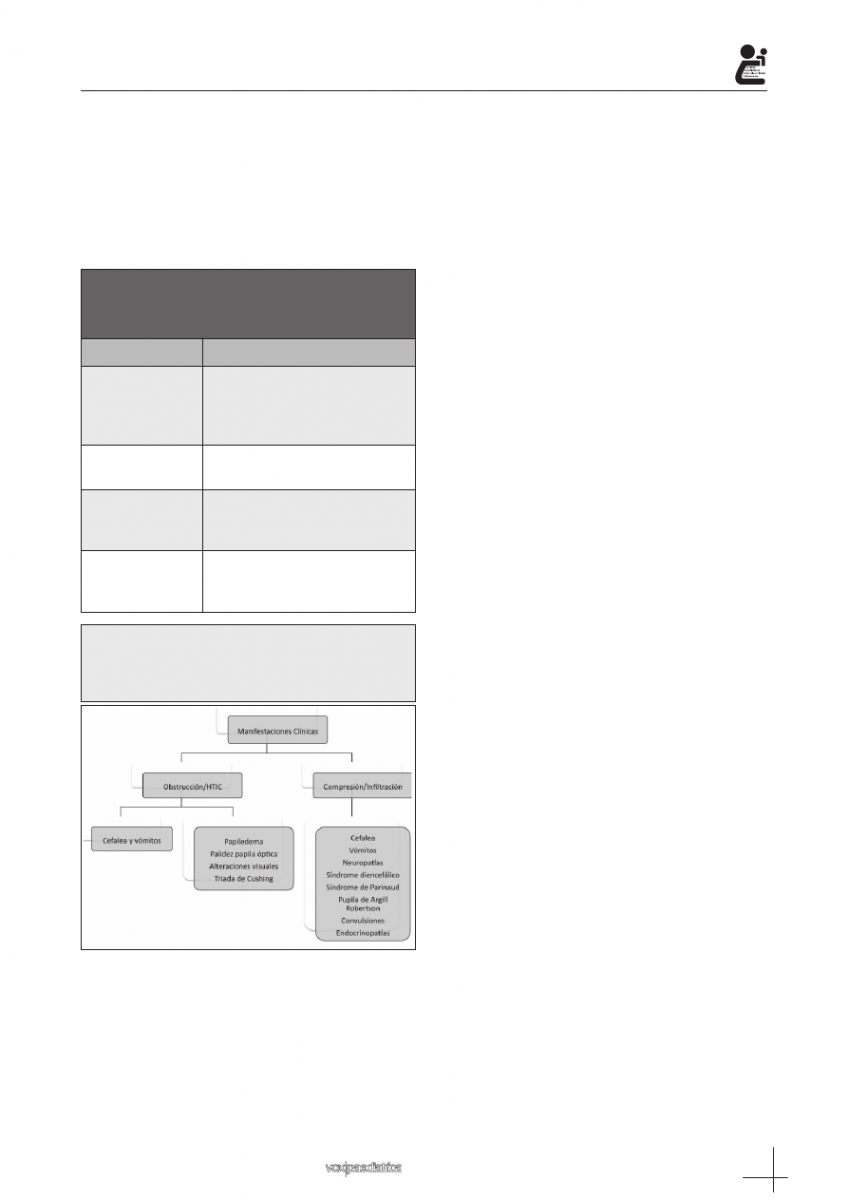
En cuanto a la clínica de presentación, los signos y
síntomas pueden agruparse según la localización del
tumor (tabla I), la existencia de obstrucción a la circu-
lación del LCR o compresión de estructuras adyacen-
tes por el mismo (figura 7).
Tabla I.: Signos y síntomas de presentación del
los tumores del sistema nerviosos central en
función de su localización.
Localización
Clínica
Supratentoriales
Cefalea, irritabilidad, convul-
siones, alteraciones lenguaje,
déficit motor y sensitivo, endocri-
nopatías
Fosa posterior
Nauseas y vómitos, cefalea,
dismetría, ataxia, nistagmo
Tronco del
encéfalo
Alteración de la marcha y
coordinación, parálisis de pares
craneales
Médula espinal
Dolor de espalda, entumecimiento
o debilidad miembros, disfunción
vesical o intestinal
Figura 7: Forma clínica de presentación de los tumo-
res del sistema nervioso central en función de la pre-
sencia de hipertensión intracraneal y/o compresión o
infiltración de estructuras adyacentes.
HTIC: hipertensión intracraneal.
Los síntomas de HTIC están presentes en el 40% de
todos los tumores intracraneales, en el 40% de los tu-
mores diagnosticados en menores de 4 años de edad
y en el 80% de los que se localizan en fosa posterior.
En un meta-análisis que incluía un total de 4171 niños
diagnosticados de tumores del SNC, la mayoría local-
izados en fosa posterior, la cefalea fue el síntoma de
presentación más frecuente, seguida de las náuseas
y vómitos, y éstos de las alteraciones de la marcha o
la coordinación
(13)
. La presencia de papiledema consti-
tuía el signo al diagnóstico más común, seguido de las
convulsiones.
En nuestra serie, el síntoma más frecuente al diag-
nóstico fue la cefalea, seguida de los vómitos, las con-
vulsiones y las alteraciones de la marcha. El signo más
común fue la presencia de papiledema. En el grupo de
niños menores de 4 años de edad, en el citado meta-
análisis la macrocefalia fue la forma más frecuente de
presentación, mientras que en nuestra serie predomi-
naron las alteraciones de la marcha. En este grupo de
edad los tumores del SNC pueden presentarse tam-
bién como cambios en el comportamiento e irritabilidad
(8). Por su parte, en los pacientes con tumor asociado
a neurofibromatosis tipo 1 la mayoría debutó con pér-
dida de agudeza visual, sin embargo, en nuestra serie
la cefalea fue el síntoma más común.
Tres de nuestros pacientes debutaron con
trastornos hormonales: uno con pubertad precoz,
otro con diabetes insípida y un tercero con diabetes in-
sípida y amenorrea. El primero se trataba de un glioma
de bajo grado de tálamo y los otros dos eran germino-
mas hipofisiarios.
Taylor y col. estudiaron retrospectivamente una co-
horte de 176 pacientes pediátricos con lesiones hipo-
tálamo-pituitarias
(14)
. La pubertad precoz se identificó
como forma de presentación de hamartomas y gliomas
de la vía óptica, mientras que la diabetes insípida con-
dujo al diagnóstico de tumores de células germinales.
En dos tercios de estos pacientes, los trastornos en-
docrinos precedieron en aparición a los signos y sínto-
mas neuro-oftalmológicos. En nuestra serie los trastor-
nos endocrinos precedieron una media de 21 meses al
resto de la clínica.
La mediana de tiempo entre el inicio de los síntomas
y el diagnóstico fue inferior en niños menores de 4 años,
dato que coincide con la literatura revisada, según la
cual la edad por encima de los 3 años es motivo de
demora diagnóstica
(12)
. Coserria y col. estudiaron ret-
rospectivamente la clínica de presentación de los tu-
mores del SNC en niños en función de edad, menores
o igual de 5 años y mayores de 5 años, y observaron
que la mediana del tiempo de demora diagnóstica era
de 50 días en el primer grupo de edad frente a 3 me-
ses en el segundo
(2)
. Coincidimos con estos autores
en que este hecho probablemente sea debido a que
en niños mayores la sintomatología se atribuye inicial-
mente a otras patologías más comunes lo cual retrasa
el diagnóstico.
En lo referente a la localización, en los niños predom-
inan los tumores infratentoriales, salvo en lactantes que
son más frecuentes los supratentoriales
(3)
. En nuestra
serie encontramos un predominio de tumores supra-
tentoriales, lo que explicamos por un mayor porcen-
taje de DNET, segundo tipo histológico más frecuente

14
S
OCIEDAD
DE
P
EDIATRÍA
DE
A
NDALUCÍA
O
CCIDENTAL
Y
E
XTREMADURA
Volumen XXI Nº 1 Mayo 2014
después del astrocitoma de bajo grado, mientras que
en la literatura publicada son más frecuentes el as-
trocitoma de bajo grado y el meduloblastoma
(1,2)
. Este
hecho justificaría también que las convulsiones como
clínica de presentación sean más frecuentes que las
alteraciones de la marcha o de la coordinación entre
nuestros pacientes, al asociarse a los DNET.
El tratamiento de los tumores del SNC debe ser mul-
tidisciplinar. La neurocirugía constituye la piedra angu-
lar con el objetivo de confirmar el diagnóstico anato-
mopatológico, aliviar la compresión que sobre estruc-
turas cerebrales ejerce el tumor y aumentar la super-
vivencia cuando la resección tumoral es completa
con márgenes libres o lo más amplia posible. Por otra
parte, por sí misma puede ser curativa en el caso de
determinados tumores como los astrocitomas de bajo
grado de fosa posterior o los craneofaringiomas. La in-
dicación de radioterapia y quimioterapia varía según la
histología del tumor, su localización, extensión y com-
portamiento biológico y la edad del paciente
(15)
Los tumores del SNC, tanto por la enfermedad en
sí misma como por la agresividad del tratamiento, en-
trañan una importante morbilidad tanto física como in-
telectual
(2)
, de ahí la necesidad de desarrollar nuevos
agentes terapéuticos que actúen selectivamente sobre
las células tumorales sin interferir en el desarrollo de las
células sanas actuando a través de las vías de señal-
ización celular implicadas en la carcinogénesis. Así, los
anticuerpos monoclonales como el nimotuzumab ac-
túan sobre los receptores del factor de crecimiento epi-
dérmico implicados en la diferenciación celular, mien-
tras que el bevacizumab inhibe al factor de crecimiento
del endotelio vascular y por consiguiente detiene la an-
giogénesis que nutre el lecho tumoral. Por otra parte,
fármacos como el everolimus inhiben la vía del m-TOR
implicada en la regulación del ciclo celular
(9)
. La admin-
istración de este último fármaco a pacientes diagnosti-
cados de astrocitomas subependimario de células gi-
gantes en el contexto del complejo esclerosis tuberosa,
se ha asociado a una disminución del tamaño tumoral y
control de las crisis convulsivas
(16)
.
Los agentes biológicos parecen relacionarse con
una menor agresividad y toxicidad que la terapia con-
vencional con cirugía, quimioterapia y/o radioterapia.
La supervivencia a los 3 años en nuestra serie es
del 90,54% y del 87,83% a los 5 años. En la literatura
revisada encontramos variaciones en función de las
distintas series. Según datos del EURCORE Working
Group, la mortalidad mostró cierta tendencia a dis-
minuir con el tiempo en la década de los 80
(17)
. La
supervivencia a los 5 años aumentó de forma significa-
tiva desde un 45% en pacientes diagnosticados en la
década de los 70 hasta un 66% en la década de los 90,
especialmente en el grupo de edad comprendido entre
los 1 y 4 años en el que el pronóstico es peor. Arndt y
col. estudiaron la supervivencia a los 10 años de niños
diagnosticados de tumores del SNC entre 1985-1999
en distintas regiones europeas
(18)
. Estimaron una su-
pervivencia del 59%, 4% superior a la de la cohorte
de niños diagnosticados entre 1985 y 1989 (55%). Ob-
servaron variaciones entre regiones geográficas, de tal
forma que la supervivencia era del 49,8% en los países
del este (Estonia, Hunfría y Eslovaquia), del 56,3% en
los del sur (Italia, Eslovenia y España), del 60,7% en
los del oeste (Francia, Alemania, Países Bsjoa, Suiza
y Reino Unido) y del 63,3% en los del norte de Europa
(Dinamarca, Finlandia, Islandia y Noruega).
En series americanas la supervivencia es mayor. Morris
y col. estudiaron la supervivencia a largo tiempo de una
cohorte de niños tratados entre los años 1985 y 2000 en
el St Jude Children´s Research Hospital (Memphis, TN),
observando que entre los pacientes que sobrevivieron al
menos 5 años la supervivencia fue del 91,3% a los 10
años y del 86% a los 15 del diagnóstico
(19)
.
Según los datos de supervivencia de base pobla-
cional recogidos en el RNTI-SEHOP, la supervivencia
a los 3 del diagnóstico oscila entre el 70% y el 78% y
alcanza el 75% a los 5 años, entre 1990 y 2004
(5)
.
En conclusión, los gliomas de bajo grado represen-
tan la variedad histológica más frecuente en niños. La
clínica de presentación de los tumores del SNC de-
pende fundamentalmente de su localización. La cirugía
continúa siendo el tratamiento de elección, asociada o
no a radioterapia y/o quimioterapia según el tipo de tu-
mor o su localización. Los nuevos agentes biológicos
constituyen una alternativa a la quimioterapia y radiote-
rapia tradicionales, con menor toxicidad, aunque son
necesarios más estudios para confirmar su eficacia y
seguridad.
Bibliografía
1. Ching Lau, Wan-Yee Teo. Epidemiology of central
nervous system tumors in children. UpToDate 2012.
http://www.uptodate.com/contents/epidemiology-of-
central-nervous-system-tumors-in-children.
2. Coserria Sá nchez JF, Garrido Ocañ a AI, Quiroga
Cantero E, Reina González AM, Amadeu Da Costa AP,
García Zara N. Clínica de presentación de los tumores
del sistema nerviosos central en función de la edad.
AnPediatr 2007;66:115-20.
3. Villarejo F, Martínez Lage JF. Tumores cerebrales
en niños. Pediatr Integral 2008;XII:557-583.
4. Fleming AJ, Chi SN. Chi. BrainTumors in Children.
Curr Probl Pediatr Adolesc Health Care 2012;42:80-103.
5. Peris Bonet R, Felipe García S, Martínez Ruíz
N, Pardo Romaguera E, Valero Poveda S. Registro
Nacional de Tumores Infantiles (RNTI-SEHOP). Cáncer
Infantil en España. Estadísticas 1980-2012. Valencia,
mayo de 2013.
6. Taylor AJ, Little MP, Winter DL, Sugden E, Ellison
DW, Stiller Ca, et al. Population-based risks of CNS
tumors in survivors of childhood cancer: the Bri-

15
Volumen XXI Nº 1 Mayo 2014
B
AENA
-G
ÓMEZ
M.A.
ET
AL
- T
UMORES
DEL
SISTEMA
NERVIOSO
CENTRAL
EN
NIÑOS
. E
XPERIENCIA
DEL
HOSPITAL
INFANTIL
REINA
SOFÍA
tish Childhood Cancer Survivor Study. J Clin Oncol
2010;28:5287-93.
7. Aristu J, Zubieta JL, Bejarano B, Narbona J, Sie-
rrasesúmaga L. Tumores del sistema nervioso central.
En Luis Sierrasesúmaga, Federico Antillón Klussmam,
ed. Tratado de Oncología Pediátrica. Enfermedades
malignas del niño y del adolescente. Primera edición.
Madrid: Pearson Educación; 2006. p.445-485.
8. Ching Lau, Wan-Yee Teo. Clinical manifestations
and diagnosis of central nervous system tumors in
children. UpToDate 2013. http://www.uptodate.com/
contents/clinical-manifestations-and-diagnosis-of-cen-
tral-nervous-system-tumors-in-children.
9. Nageswara Rao AA, Scafidi J, Wells EM, Packer
RJ. Biologically Targeted Therapeutics in Pediatric
Brain Tumors.PediatrNeurol 2012;46:203-11.
10. Marks AM, Packer RJ. A review of secondary
central nervous system tumors after treatment of a
primary pediatric malignancy. SeminPediatrNeurol
2012;19:43-8
11. Neglia JP, Robison LL, Stovall M, Liu Y, Packer
RJ, Hammond S, et al .New primary neoplasms of
the central nervous system in survivors of childhood
cancer: a report from the Childhood Cancer Survivor
Study. J NatlCancer Inst 2006;98:1528-37.
12. Pinho RS, Andreoni S, Silva NS, Cappellano AM,
Masruha MR, Cavalheiro S, et al. Pediatric central ner-
vous system tumors: a single-center experience from
1989 to 2009. J Pediatr Hematol Oncol 2011;33:605-9.
13. Wilne S, Collier J, Kennedy C, Koller K, Grundy
R, Walker D. Presentation of childhood CNS tumours:
a systematic review and meta-analysis. Lancet Oncol
2007;8:685-95.
14. Taylor M, Couto-Silva AC, Adan L, Trivin C,
Sainte-Rose C, Zerah M, et al. Hypothalamic-pituitary
lesions in pediatric patients: endocrine symptoms often
precede neuro-ophthalmic presenting symptoms. J
Pediatr. 2012;161:855-63.
15. López-Aguilar E, Sepúlveda-Vildósola AC, Rios-
covian-Soto AP, Pérez-Ramírez JD, Siordia Reyes G.
Tumores cerebrales en pediatría. Estado actual del
diagnóstico y tratamiento. GAMO 2011; 10:41-45.
16. Krueger DA, Care MM, Holland K, Agricola K,
Tudor C, Mangeshkar P, et al. Everolimus for subepen-
dymal giant-cell astrocytomas in tuberous sclerosis. N
Engl J Med. 2010;363:1801-11.
17. Magnani C, Aareleid T, Viscomi S, Pastore
G, Berrino F; EROCARE Working Group. Variation
in survival of children with central nervous system
(CNS) malignances diagnosed in Europe between
1978 and 1992: the EUROCARE study. Eur J Cancer
2001;37:711-21.
18. Arndt V, Kaatsch P, Steliarova-Foucher E, Peris-
Bonet R, Brenner H. Up-to-date monitoring of child-
hood cancer long-term survival in Europe: central
nervous system tumors. Ann Oncol 2007;18:1734-42.
19. Morris EB, Gajjar A, Okuma JO, Yasui Y, Wallace
D, Kun LE, et al. Survival and late mortality in long-
term survivors ps pediatric CNS tumors. J Clin Oncol
2007;25:1532-8.
| Adjunto | Tamaño |
|---|---|
| voxpaed21.1pags9-15.pdf | 291.18 KB |
Síguenos en


© 2003-24 Sociedad de Pediatría de Andalucía Occidental y Extremadura. Editor de la web: Jaime J. Cuervo Valdés. | Actualizada el 27 de diciembre de 2024 |